


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
 molecule adsorption on polar and
molecule adsorption on polar and 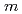 -plane surfaces of GaN
-plane surfaces of GaNSumiya, Masatomo*; Sumita, Masato*; Asai, Yuya*; Tamura, Ryo*; Uedono, Akira*; Yoshigoe, Akitaka
Journal of Physical Chemistry C, 124(46), p.25282 - 25290, 2020/11
Times Cited Count:11 Percentile:47.05(Chemistry, Physical)The initial oxidation of different GaN surfaces [the polar Ga-face (+c) and N-face (-c) and the nonpolar (10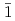 0) ()plane] under O molecular beam irradiation was studied by real-time synchrotron radiation X-ray photoelectron spectroscopy and DFT molecular dynamics calculation. The results predict that triplet O either dissociates or chemisorbs at the bridge position on the +c-surface, while on N-terminated -c-surface the O
0) ()plane] under O molecular beam irradiation was studied by real-time synchrotron radiation X-ray photoelectron spectroscopy and DFT molecular dynamics calculation. The results predict that triplet O either dissociates or chemisorbs at the bridge position on the +c-surface, while on N-terminated -c-surface the O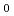 2 molecule only undergoes dissociative chemisorption. On the -GaN surface, although the dissociation of O is dominant, the bond length and angle were found to fluctuate from those of O molecules adsorbed on the polar surfaces. The computational model including both the surface spin and polarity of GaN is useful for understanding the interface between GaN and oxide layers in metal-oxide electronic.
2 molecule only undergoes dissociative chemisorption. On the -GaN surface, although the dissociation of O is dominant, the bond length and angle were found to fluctuate from those of O molecules adsorbed on the polar surfaces. The computational model including both the surface spin and polarity of GaN is useful for understanding the interface between GaN and oxide layers in metal-oxide electronic.
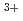 ion; A Car-Parrinello molecular dynamics study
ion; A Car-Parrinello molecular dynamics studyIkeda, Takashi; Hirata, Masaru; Kimura, Takaumi
Journal of Chemical Physics, 122(2), p.024510_1 - 024510_5, 2005/01
Times Cited Count:31 Percentile:70.44(Chemistry, Physical)The solvation shell structure of Y and the dynamics of the hydrated ion in an aqueous solution of 0.8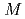 YCl
YCl are studied in two conditions with and without an excess proton by using first principles molecular dynamics method. We find that the first solvation shell around Y contains eight water molecules forming a square antiprism as expected from X-ray absorption near edge structure in both the conditions we examined. A detailed analysis relying upon localized orbitals reveals that the complexation of water molecules with yttrium cation leads to a substantial amount of charge redistribution particularly on the oxygen atoms, giving rise to the chemical shifts of
are studied in two conditions with and without an excess proton by using first principles molecular dynamics method. We find that the first solvation shell around Y contains eight water molecules forming a square antiprism as expected from X-ray absorption near edge structure in both the conditions we examined. A detailed analysis relying upon localized orbitals reveals that the complexation of water molecules with yttrium cation leads to a substantial amount of charge redistribution particularly on the oxygen atoms, giving rise to the chemical shifts of  -20 ppm in
-20 ppm in 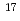 O NMR relative to the computed nuclear shieldings of the bulk water.
O NMR relative to the computed nuclear shieldings of the bulk water.
 molecular dynamics study of polarization effects on ionic hydration in aqueous AlCl solution
molecular dynamics study of polarization effects on ionic hydration in aqueous AlCl solutionIkeda, Takashi; Hirata, Masaru; Kimura, Takaumi
Journal of Chemical Physics, 119(23), p.12386 - 12392, 2003/12
Times Cited Count:43 Percentile:78.85(Chemistry, Physical)The solvation shell structure and dynamics of Al and Cl in an aqueous solution of 0.8 M AlCl are studied under ambient conditions by using
in an aqueous solution of 0.8 M AlCl are studied under ambient conditions by using  molecular dynamics method. The solvation structures obtained from our simulations are in good agreement with the experimental ones for both Al and Cl. A detailed analysis of intramolecular geometry of hydration waters and dipole moments of the ingredients shows that the polarization has substantial effects on the structures and dynamics of both the cation and anion hydration shells. Implications to the metal hydrolysis of Al will also be given.
molecular dynamics method. The solvation structures obtained from our simulations are in good agreement with the experimental ones for both Al and Cl. A detailed analysis of intramolecular geometry of hydration waters and dipole moments of the ingredients shows that the polarization has substantial effects on the structures and dynamics of both the cation and anion hydration shells. Implications to the metal hydrolysis of Al will also be given.
Machida, Masahiko; Nakamura, Hiroki
no journal, ,
